DNA合成常見問題與分析
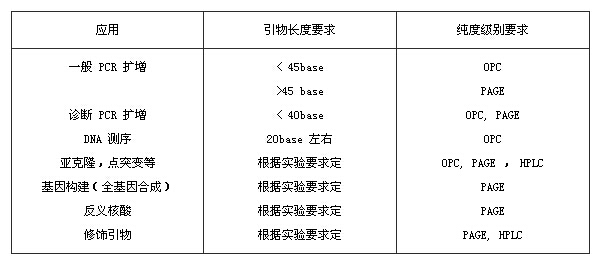
1. 引物純化方式有哪些,如何選擇?
? ? C18柱脫鹽:這是一種活性碳柱子,有人稱其為簡易反相柱,它對DNA有特異性的吸附,DNA可以被有機溶液洗脫,但不會被水洗脫,所以能有效地去除鹽分。但是它不能有效去除比目的片段短的小片段。
? ? OPC:合成DNA寡核苷酸片段(Oligo)的最終產物上帶有DMT。OPC純化就是基于DMT與樹脂的親合力。Oligo上只有最后一個堿基帶有DMT,其它堿基的DMT基團在逐步的合成過程中已被去除。因此,原則上只有真正所要合成的DNA片段末端有DMT,而短片段雜質末端無DMT。
? ?OPC柱中裝有對DMT具有親和力的樹脂,所有合成產物吸附在OPC柱上以后,用稀的有機溶劑洗柱,帶有DMT的片段吸附能力強,不易被洗脫,不帶有DMT的片段吸附能力弱,被洗脫。然后用三氟乙酸TFA脫去DMT基團,再用濃一點的有機溶劑洗脫DNA。這種方法的優點是快速,簡易。但是其專一性吸附DMT能力有限,不免仍然有短片段帶入的可能,特別是對長堿基的純化效果不一定理想。長片段建議使用PAGE純化。
? ? PAGE:它是依據DNA片段在變性聚丙烯酰胺凝膠中電泳時的遷移率不同來分離大小片段的。由于各分子所帶電荷和大小不同,綜合影響其在凝膠中的遷移速度,大片段遷移得慢,經過一定時間的電泳,大小片段會分開,然后停止電泳,剝離凝膠,置于熒光TLC板上在紫外燈下切割目的條帶,浸泡碎膠,并從泡膠的鹽溶液中回收目的DNA。優點是純化效果很好、尤其是純化長鏈效果更好、而且是可以直觀看見DNA片段合成情況的質控環節。缺點是費人工、電泳及后處理過程中樣品損失量大。
2.需要什么級別的引物?
3. 需要合成多少OD數?
? ? 根據實驗目的確定。一般PCR擴增,2OD引物,可以做200-500次50ul標準PCR反應。如果是做基因拼接或退火后做連接,1OD就足夠了。但是有些研究人員,就做幾次PCR,卻要5-10OD。做全基因構建的引物都比較長,但是我們有些研究人員也要求高OD數。片段越長,最后全長得率就越低,割膠越多,出錯的幾率就越大。超出需要之外的OD數要求,其實也是對社會資源的一種浪費,同時也從一個側面反映了部分研究人員,特別是新手的自信心不足。引物用量過大,容易引起非特異性擴增。
4.?投訴定量不準是怎么回事兒?
? ? 可能存在如:定量錯誤,分裝問題,系統誤差等(10%左右是允許的)現象,而客戶投訴的大部分原因是:因為沒有能夠正確理解引物OD數的含義,沒有能夠正確使用分光光度計,特別是使用微量測定;用戶沒有將OD讀數,正確地轉換成母液中OD數。這種情況比較常見。例如:驗證標有2OD引物的簡單做法是:加入1ml水,徹底溶解混勻后,取100ul,加入裝有900ul水,光徑為1cm的石英比色皿中操作方法好象不對,波長260nm,此時光吸收的讀數為0.2。其他情況如用戶收到引物干粉時,打開引物管蓋前沒有離心或其他誤操作導致引物干粉部分丟失。
5.?如何計算引物的濃度?
? ? 引物保存在高濃度的狀況下比較穩定。引物一般配制成50-100pmol/ul。溶解前您需要核對合成報告單和引物標簽上的引物OD數是否一致。如果不一致,請和我們聯系。我們可以根據生產記錄查到實際產量是多少。
? ??一般情況下,我們建議將引物的濃度配制成100umol/L,加水的體積(微升)按下列方式計算:V(微升)=OD數*(乘)30*(乘)1000/(除)引物的分子量。引物的分子量可以從合成報告單上獲得。如果需要配制成其他濃度,按上述公式換算。
? ? 注意:1OD260=30ug/ml
6.?如何計算引物的Tm值?
? ?引物設計軟件都可以給出Tm,Tm與引物長度、堿基組成及所使用緩沖液的離子強度有關。
? ??長度為25mer以下的引物,Tm的近似計算公式為:Tm=4℃(G+C)+2℃(A+T)
? ? 對于更長的寡聚核苷酸,Tm計算公式為:
? ? Tm=81.5+16.6xLog10[一價陽離子]+0.41(%GC)–600/size
? ? 公式中,Size=引物長度。
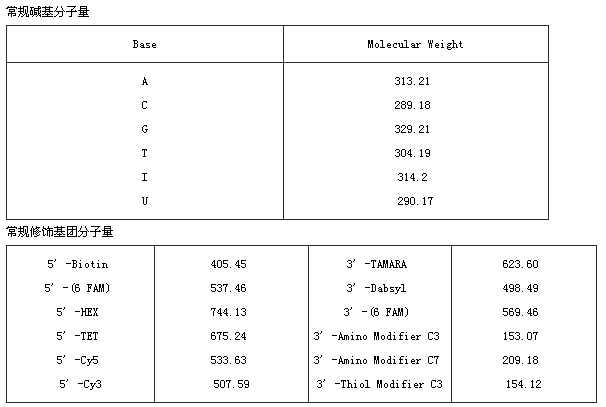
7.?引物(含修飾)的分子量是如何確定的?
? ??MW=(NA*WA)+(NC*WC)+(NG*WG)+(NT*WT)+(Nmod*Wmod)+(Nx*Wx)+(Ni*Wi)+16*Ns–62
? ? NA,NG,NC,NT,Ni分別為引物中堿基A或G或C或T或I的數量,WA,WC,WG,W,Wi分別為引物中堿基A或G或C或T或I的分子量,Nmod,Wmod分別為修飾基團的數目和分子量
? ? 對于混合堿基的分子量為混合堿基的分子量總合除以混合數,例如G+A混合的分子量為(313.21+329.21)/2 =321.21。Ns為硫代數目,硫代每個位置增加分子量16
8. Oligo DNA最長可以合成多少個堿基?
? ??以每步反應效率為99%進行計算,粗略計算合成到100bp時,目的DNA片段的比例便為37%。建議合成長度在100mer以內,比較長的的DNA片段,建議在本公司的基因合成部合成。
9. 寡核苷酸一般以何種形式提供?如何溶解?
? ? 我們一般以干粉形式提供寡核苷酸,呈白色或無色透明狀。
建議使用TE buffer溶解,如果實驗條件限制,也可以使用雙蒸水或者去離子水等不影響后續實驗的緩沖液。
10. 寡核苷酸在使用時有哪些問題需要注意?怎樣保存?
? ? 我們提供的寡核苷酸是干粉形式的,有時會離開管底,因此在第一次開蓋前離心或者將管底朝下在桌子上輕叩幾次,將其收集到管子底部。
? ? 干粉狀的DNA在-20攝氏度 可以保存1年之久,但溶解后的保存時間要大大縮短。保存時間與所用的水中有無核酸酶、PH值和環境溫度等外在條件有密切的關系。我們公司一般默認提供兩管引物,建議先將一管配成較高濃度的引物溶液,置于-20攝氏度 的保存環境中,可以存放數周,這樣可以避免因反復凍融而造成的污染與破壞。
11. 引物溶解后發現有微量沉淀,會影響實驗結果嗎?
? ??純化時偶爾會有微量的樹脂進入純化好的產品中,不影響任何反應結果,請稍許離心后取上清使用。
12. 引物檢測方法
? ? 建議使用分辨率比較高的聚丙烯酰胺凝膠來檢測(濃度16%左右)。檢測后剝離凝膠,置于熒光TLC板上在紫外燈下觀察條帶的分布或者亮度。也可以用EB染色后觀察,但是EB等染色劑的染色是嵌入DNA的雙鏈之間的,而合成的Oligo DNA是單鏈,Oligo DNA自身形成的立體結構越復雜,EB的染色就越容易,DNA帶也就越亮。相反,有一些Oligo DNA由于不形成高級結構,根本就不為EB所染色,導致條帶弱甚至是無條帶,可能會出現EB染色后不能觀測到或者DNA跑不開,故不建議使用瓊脂糖凝膠電泳檢測引物純度與量。
13. 已經溶解的引物,為什么原先使用正常,而過一段時間再使用就不好了?
? ? 如果您溶解引物的水的PH值過低或污染了菌或核酸酶,會使引物降解。使用時沒有充分解凍混勻,液體不均勻也可能會造成引物加入量不準確。建議分裝引物,避免反復凍溶。建議使用10mM Tris(pH=7.5)的緩沖液溶解引物,因為有些蒸餾水的pH值比較低(pH4-5),引物在這種條件下不穩定。還有一種可能性是引物沒有問題,而是PCR使用材料特別是模板的質量與先前使用的不完全一致。
14. 引物是經過PAGE純化的,為什么還有堿基缺失或插入?
? ??理論上分析型PAGE變性電泳,可以區分引物之間一個堿基的差別。但是制備PAGE電泳,上樣量都是非常大,電泳時的條帶非常寬,帶與帶之間有重疊,分辨率已下降,電泳后割帶回收目的引物時,很難說不割到差別僅幾個堿基的引物。
15. 一般的合成OligoDNA的5和3末端有磷酸根嗎?
? ? 沒有,5’和3’末端均為羥基。如需要加磷酸末端,訂貨時請特別注明,需另收取費用。
16. 簡并引物使用應該注意什么?
? ? 簡并引物是指代表編碼單個氨基酸所有不同堿基可能性的不同序列的混合物,次黃嘌呤可以同所有的堿基配對,降低引物的退火溫度。
? ? 使用時注意
? ??①不要在引物的3 '端使用簡并堿基,最后三個堿基的退火溫度足以在錯誤位點起始PCR
? ? ②使用稍高的引物濃度(1uM-3uM),許多簡并混合物中的引物不是特異性針對模板的
? ? ③簡并引物應選用簡并程度低的密碼子
? ??④3'端不應存簡并性,否則可能出現產量低而看不見擴增產物
? ? ⑤最好不用來測序
17. PCR產物只有單引物形成的非特異性條帶?
? ? 引物特異性不好,建議重新設計引物。
18. 檢測PCR產物時,經常看到下面有很多明顯的引物二聚體形成的亮帶?
? ? 可以通過減少引物的用量或者減少循環次數來避免引物二聚體產生,如果這樣做效果還不明顯,再考慮引物的問題,說明引物之間容易形成互補的配對結構。
19. 引物設計參考
? ? ①引物的3’最好是C或者G,或者CG,GC,能促進引物通過G與模板結合。
? ? ②設計的引物最好有接近的Tm值
? ? ③避免引物3’互補或者自身互補
? ??④避免引物3’有三個G或者C,避免在擴增高CG含量的模板時產生錯配引物的退火溫度主要是指引物與模板退火的溫度,不包括酶切位點
20. 引物是我們設計的,現在您擴不出來,我們要負責嗎?
? ? 不負責任。引物設計好,我們都會發給您確認。設計引物時我們遵循一般的指導原則,由于影響后期實驗的因素很多,因此,我們不負責后續續問題,但可以根據客戶要求重新設計。
21. 合成的引物進行 PCR 反應時無目的條帶,怎么辦?
? ? PCR反應失敗的原因很多,可以從以下幾個方面考慮。
? ? ①引物和模板是否配對,同源性有多大?
? ? ②引物本身是否有高級結構,或者二條引物之間是否形成高級結構?
? ??③PCR反應所用試劑是否能正常工作?
? ? ④PCR儀是否能正常工作?
? ? ⑤PCR反應條件是否合適?
? ? 如果一切正常,還無法解決問題時,我們會重新檢測引物,必要時免費重新合成。
22. PCR擴增有很強的非特異條帶,說明引物有污染嗎?
? ? 擴增目標很弱或沒有,非特異性條帶很亮,一般是模板污染(如RNA中污染基因組)或擴增條件不合適所致,換模板或者改變擴增條件,如降低退火溫度等。如果有主帶,還有非特異性條帶,需要適當提高退火溫度。
23. PCR產物克隆后測序發現引物堿基有錯誤,或是缺失突變堿基,為什么?
? ? ①合成過程中,堿基附加至DNA片段之前,堿基和堿基之間發生了偶聯,然后再附加到了DNA片段上,造成多堿基現象。
? ? ②合成時堿基G可能會轉化成烯醇異構體,此時進行PCR反應時G會轉變成A。
? ? ③合成過程中的脫嘌呤作用,對脫嘌呤后的堿基DNA聚合酶不能識別,此時可能觀察到A或G的缺失。嘌呤含量越高,就越容易發生這種情況。
? ??④不完全的脫保護結果。通常C脫的快,G脫的慢。不完全脫保護會發生堿基缺失。
? ? ⑤合成過程中的Capping(蓋帽)反應不完全,使短片段DNA繼續參與合成,造成堿基缺失。
? ? ⑥2,6 diaminopurine , DNA復制和擴增過程中DNA聚合酶將2,6 diaminopurine看作堿基A,測序就會發現堿基G-A置換。
? ? 應該說,上述這些情況發生的可能性都極低。合成的DNA鏈越長,發生這種情況的機率也就越大。多數問題的出現是PCR過程和克隆過程中引入的錯誤。如果出現這種情況,你可以,重新挑取克隆測序,有可能找到序列正確的克隆。可以要求我們重新免費合成引物 ,我們不承擔其他連帶責任。
24. 為什么我們的引物重合了幾遍都擴增不出來?
? ? 如果您懷疑引物的問題,請您首先測定您溶解的引物的OD值,看實驗時加入的引物量是否正確。如果量是正常的,請您告訴我們公司您的引物編號,我們會復查留存樣品。如不明原因,我們免費為您重新合成一次。如果仍然不能擴增,請您查找其它原因,很可能是酶或者條件不夠成熟。
? ? OLA技術是通過兩步來完成的,先用PCR技術進行擴增,再將PCR產物變性為單鏈;然后加入兩條互補于待測SNP位點一側序列的等位基因特異性探針和一條互補于待測SNP位點另一側序列的公共探針,在DNA連接酶的作用下,與目標DNA?單鏈完全互補的等位基因特異性探針和公共探針發生連接反應。
25. 長鏈引物為什么出錯的幾率非常高?
? ? 引物合成時,每一步反應效率都不能達到100%,產生堿基插入,缺失,置換突變的因素客觀條件都有一直存在。引物鏈越長,突變的頻率累加起來就越高。研究人員總希望合成的引物萬無一失,這種心情可以理解。但是猶如PCR擴增,不可能絕對保證擴增產物中沒有突變,引物合成也不可能保證100%正確。要知道,引物合成中發生錯誤(非人為因素)的頻率,比任何高保真高溫聚合酶PCR擴增過程所產生的頻率都要高。做長鏈引物合成,您要有引物中部分引物可能有突變的思想準備。
26. PCR擴增不出就引物有問題嗎?
? ? 基本不是。當今發展的各種的PCR擴增技術,各種的高溫聚合酶,就是來解決PCR擴增中遇到的擴不出,擴增效率低的問題。如巢式PCR就是擴增那些拷貝數很低的基因片段。 有些重復片段的擴增,GC含量高的片段,非要采用特殊擴增手段才能擴增出來。
? ? 引物擴增不出,主要是下列兩種情況比較常見
? ? ①RT-PCR:請注意,很多基因通過常規RT–PCR方法是很難擴增出來的。RT-PCR成功的關鍵在于RT反應的RNA質量和目標基因在特定組織和細胞中的含量。
? ??②從基因組中擴增:一般情況下,基因在基因組中都是單拷貝,基因組作為模板需要嚴格控制用量,基因組DNA過高,會影響反應體系中的Mg和pH。
27. 測序發現引物有突變是怎么回事?
? ? 測序發現引物區域有突變,特別是40個堿基以下的引物,發生的概率不大,但是肯定也會發生。用戶一般可以放心,引物序列一般都是通過電腦直接將您的序列COPY到合成儀的,堿基輸錯的機會不多。我們有一套控制辦法,預防堿基輸入錯誤。發生這種突變的原因有很多解釋,人們還沒有辦法徹底解決這個問題。引物合成的固相合成原理都一樣,采用的機器也基本相同,合成主要原料都是由少數的幾家跨國公司提供的,所有每個合成服務商遇到的問題也基本類似,沒有人可以超脫。
? ? 引物合成是一種多步驟的化學反應,合成效率最高也就是99%,副產品不可能避免。引物序列中插入突變往往是堿基重復,一般認為,偶聯過程中,正在偶聯的部分單體發生丟失DMT,導致單體又接了上去,故發生插入同一堿基的突變。至于缺失突變,一般認為是帶帽(capping)反應不徹底造成的,Caping反應主要是封閉極少數5'-羥基沒有參加反應單體。被封閉的引物,在下一輪偶聯時將不能繼續參與合成。對于堿基置換的突變,產生的原因一般認為是堿基不能100%脫保護,即引物上可能含有殘留保護基團,引物的這些區域不能很好地與互補鏈配對,當擴增的產品被亞克隆轉化到大腸桿菌中,可能被細菌中修復系統補上了非配對的堿基。置換突變通常發生在G轉換成其它堿基。堿基G在一定條件下可以轉化為烯醇異構體(脫嘌呤),2,6diaminopurine,DNA復制和擴增過程中DNA聚合酶將2,6 diaminopurine看作堿基A,測序就會發現堿基G-A置換。脫嘌呤現象在富含嘌呤的引物中發生的頻率較高。脫嘌呤的引物在引物后處理脫保護階段如果被降解,測序就會發現堿基G或A的缺失。